
Whether you want to investigate gene functions or explore genetic variations, extracting a gene or another subsequence from a genome assembly is a common task that you will often be doing. In this post, we will explore how we can easily extract a gene from a FASTA file by using its coordinates in a GFF file. In this particular case, we will use the publicly available genome assembly and annotation of Arabidopsis thaliana.
Setup
Software
For this, you will need Python as well as the Biopython and argparse packages. Once Python is installed on your system, you can easily get them with:
python3 -m pip install bio argparse
or with this line if you are not an administrator of your system (the --user flag is used to install the package only for your user):
python3 -m pip install --user bio argparse
Input files
To download the genome assembly and annotation of Arabidopsis thaliana, visit the NCBI datasets website and click on the Download button. Be sure to select Genome sequences (FASTA) and Annotation features (GFF) before clicking on Download at the bottom of the popup.
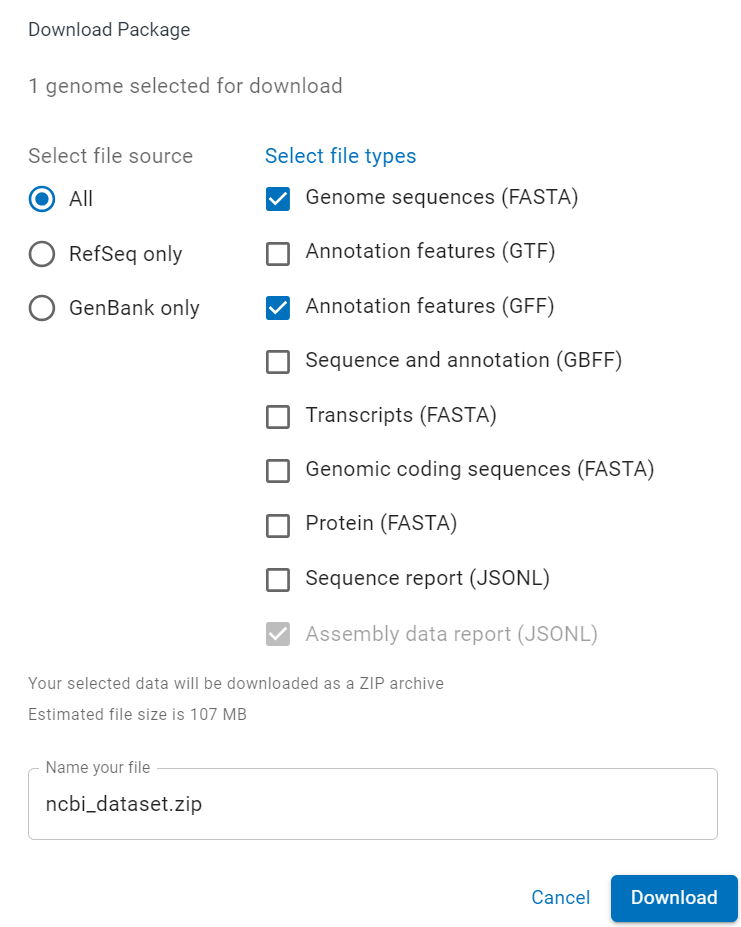
Once the file is downloaded, simply unzip it somewhere so you can follow along. Inside the data folder, you will find two folders named GCA_000001735.2 and GCF_000001735 (at the time of writing, yours may be different). The GCA one contains the Genbank assembly while the GCF one is the RefSeq assembly. There is no sequence difference between the two (take a look at this if you want to know more about it) so we will go with the RefSeq assembly.
Coding our solution
Goal of the project
Before starting to code anything, it is important to set goals so you can be prepared for what you have to code. Here we want to extract a single gene from a genome assembly so our code will have 4 inputs:
- the path to the assembly we want to extract the gene from
- the name of the sequence the gene is on
- the beginning of the gene
- the end of the gene
Example data
By looking at the GFF file (GCF_000001735.4/genomic.gff) I selected a gene at random, specifically this one:
NC_003070.9 RefSeq gene 51953 54737 . + . ID=gene-AT1G01110;Dbxref=Araport:AT1G01110,TAIR:AT1G01110,GeneID:839394;N
ame=IQD18;gbkey=Gene;gene=IQD18;gene_biotype=protein_coding;gene_synonym=IQ-domain 18,T25K16.10,T25K16_10;locus_tag=AT1G01110
This line tells us that the gene gene-AT1G01110 is located on the NC_003070.9 chromosome, it starts at coordinate 51953 and ends at 54737. Now that we know that, let’s get to work!
Command line interface
We want to make things as clean as we can so we will design a nice way for the user to interact with our program. To do this, we will use the argparse python package which provides a neat way to get parameters from the command line.
First, import the python package and declare our new program:
import argparse
# This code will not be executed if another script tries to import it
# It is good practice to include it in our main script
if __name__ == "__main__":
parser = argparse.ArgumentParser(description="Extracts a subsequence from a FASTA file.")
Now that we have a description set, let’s go back to the Goal of the project part and remember that we wanted four input parameters. We’ll add these parameters and set them as required. We will have a -g for the input genome, -n for the name of the sequence that contains our gene, -s for the start coordinate and -e for the end coordinate:
parser.add_argument("-g", action="store", dest="genome", required=True, help="Path to an input genome in FASTA format")
parser.add_argument("-n", action="store", dest="name", required=True, help="Name of the sequence the subsequence is located on")
parser.add_argument("-s", action="store", dest="start", required=True, help="Start coordinate")
parser.add_argument("-e", action="store", dest="end", required=True, help="End coordinate")
Finally, parse the command line parameters and save the result to a variable name args:
args = parser.parse_args()
Sanity checks
We want to be extra careful with user-provided data and so we will check that everything looks correct before trying to extract anything. Here is what we will check:
- does the input genome exist?
- is it readable?
- are the coordinates valid? Are start or end less than 0? Is end inferior to start?
Let’s first check that the file exists and is readable with the os module. We will crash if the input file is invalid. Add an import to the top of your file:
import os
import sys
and this code after the arguments parsing:
# Check that the genome exists and is readable
if not os.path.exists(args.genome):
print("Input genome file does not exist")
sys.exit(1)
elif not os.access(args.genome, os.R_OK):
print("You do not have the permissions to read the input genome file")
sys.exit(1)
Then, we want to verify that coordinates are numeric and valid:
# Check that start and end are numeric
try:
args.start = int(args.start)
args.end = int(args.end)
except:
print("Conversion error: '-b' and '-e' should be numeric")
sys.exit(1)
if args.end < args.start:
print("End coordinate should be superior to the the start coordinate")
sys.exit(1)
if args.start < 0 or args.end < 0:
print("Start and end coordinates should be superior to 0")
sys.exit(1)
Extracting the gene
Now that we checked that everything looks okay, we can finally try to extract our gene. To do that, we will parse all sequences in the input genome. If the name of the sequence does not match, we skip it. Otherwise, we extract the sequence that goes from start to end, without forgetting to check that end is inferior to the sequence size (the user could have mistyped something!).
First, import the Biopython module:
from Bio import SeqIO
Then, open the file in read mode and parse the sequences:
with open(args.genome) as genome:
for record in SeqIO.parse(genome, "fasta"):
Skip unwanted sequences:
# Skip the sequence if the ID is not the one we are looking for
if record.id != args.name:
continue
Check that the end is valid if the sequence is the one we want or crash otherwise:
# Check that end is not superior to the sequence length
if args.end > len(record.seq):
print(f"End coordinate is superior to sequence length: {args.end} > {len(record.seq)}")
sys.exit(1)
If everything looks good, we extract the part of the sequence that we want, print it and break out of the loop:
gene = SeqRecord(record.seq[args.start:args.end], id="gene")
print(gene.format("fasta"))
break
Our program is now complete, you can test it with (the > sign is to redirect the output of the program to a file of our choice):
python3 name_of_your_script.py -n NC_003070.9 -g GCF_000001735.4_TAIR10.1_genomic.fna -s 51953 -e 54737 > AT1G01110.fasta
Full code
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
import argparse
import os
import sys
if __name__ == "__main__":
parser = argparse.ArgumentParser(description="Extracts a subsequence from a FASTA file.")
parser.add_argument("-g", action="store", dest="genome", required=True, help="Path to an input genome in FASTA format")
parser.add_argument("-n", action="store", dest="name", required=True, help="Name of the sequence the subsequence is located on")
parser.add_argument("-s", action="store", dest="start", required=True, help="Start coordinate")
parser.add_argument("-e", action="store", dest="end", required=True, help="End coordinate")
args = parser.parse_args()
# Check that the genome exists and is readable
if not os.path.exists(args.genome):
print("Input genome file does not exist")
sys.exit(1)
elif not os.access(args.genome, os.R_OK):
print("You do not have the permissions to read the input genome file")
sys.exit(1)
try:
args.start = int(args.start)
args.end = int(args.end)
except:
print("Conversion error: '-b' and '-e' should be numeric")
sys.exit(1)
if args.end < args.start:
print("End coordinate should be superior to the the start coordinate")
sys.exit(1)
if args.start < 0 or args.end < 0:
print("Start and end coordinates should be superior to 0")
sys.exit(1)
with open(args.genome) as genome:
for record in SeqIO.parse(genome, "fasta"):
# Skip the sequence if the ID is not the one we are looking for
if record.id != args.name:
continue
# Check that end is not superior to the sequence length
if args.end > len(record.seq):
print(f"End coordinate is superior to sequence length: {args.end} > {len(record.seq)}")
sys.exit(1)
gene = SeqRecord(record.seq[args.start:args.end], id="gene")
print(gene.format("fasta"))
break
{kind=link}